Toolbar and window functionality
Selection of signature words
Report panel
Word filtering
Word search and browsing
Database info and help
Command line utilities
Publications
Here we want to present a database of 172,636 selected 8 to 14mer oligonucleotides, which frequencies varies significantly in different bacterial genomes. The database is supplied with a GUI to allows database browsing and selection of the best discriminative words for a given set of genomes or taxonomic units. A database file is available for download containing the information about frequencies of signature words in 724 bacterial genomes. Users may add new genomes to the database or remove them from the database to keep it updated and focused on the species of interest. The program was developed for comparative genomics and binning unknown sequences or groups of sequences to bacterial taxonomic units.
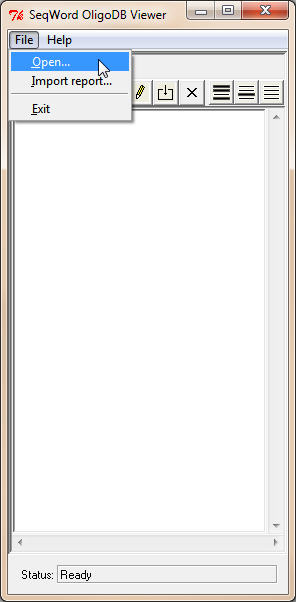 The program
OligoDBViewer is written on Python and needs
Python ver. 2.5.4
to be installed on the machine. GUI interface is based on
Pmw megawidgets. This module is
included in the download file.
The program
OligoDBViewer is written on Python and needs
Python ver. 2.5.4
to be installed on the machine. GUI interface is based on
Pmw megawidgets. This module is
included in the download file.Download the file OligoDBViewer.zip from the site http://www.bi.up.ac.za/SeqWord/downloads/. Unzip file to a selected directory. A folder OligoDBViewer will appear with several files and subordinate folders inside. Run Python OligoDBViewer.exe.py. A starting window of the program will appear.
Use the command File->Open to open a database file. The viewer cannot create a new empty database. An example of a small database example.wdb (35.1 Mb, 174 bacterial genomes) you may find in the subfolder db. Select this file and click Open. You may download the database file bacteria.wdb. At the time of writing this document, the database size was 118 Mb and it contained 733 bacterial genomes. However, this database is regularly updated and the real size of the database file may be bigger.

1. List of
taxonomic units
The list of taxonomic units is three levels deep: classes, genera and
genomes (the latter corresponds to a chromosome with a specific NC accession
number).
Multiple items in the list may be selected simultaneously by mouth clicking.
To open a taxonomic unit, double-click on it. Double-click again to collapse
the list of subunits.
2. Toolbar
▪
Open – open another database file, the
same as File->Open;
▪ Save – save
changes in the database, same as File->Save
or File->Save as;
▪ Find – open
a dialog Search genome that allows
finding a genome by its taxonomy or NC accession number. You may choose Edit->Search.
▪ Edit – open a
dialog to edit the active (last clicked) taxon as shown below. Alternatively choose Edit->Edit.

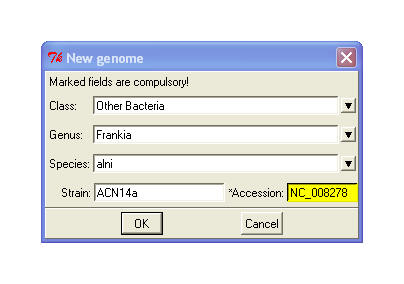
▪
New taxon – first asks for the file in
FASTA or GenBank format with the genome sequence (alternatively choose Edit->Add). Program automatically
calculates frequencies of the signature words in a given genome. Next a
dialog pops up (shown right) where class, genus, species, strain and accession
must be filled in. If a GenBank (.gbk) file was provided, program tries to
identify the phylogeny of the organism basing on the stored data. The
process of word frequency calculation is time and computer memory consuming.
It'd better to run it on a remote server using a command line utility
dbupdate_cmd.py.
▪ Delete –
remove the selected taxon from the database, same as
Edit->Delete.
Next three buttons Select all,
Reverse selection and
Deselect all operate selecting of items in
the list.
Users may create own databases by saving the existing database under a new name followed by adding and removing genomes in the databases. Two existing database then may be merged by the command File->Merge database.
1.
Selection of diverse words.
First select in the list the taxonomic units you want to compare. The
program will analyze frequencies of words in all genomes belonging to these
units. User may select units of different levels, for example: one class and
several genera of another class. Having all taxonomic units selected, click
the button Select diverse words or
choose Command->Select
diverse words. A filter setting dialog will pop up (will be discussed later,
at this stage it is not recommended to use the filter as it will
significantly slow down the program). Click OK and a list of
selected words (100 by default) with the highest discriminative power will
be shown on the right report panel.
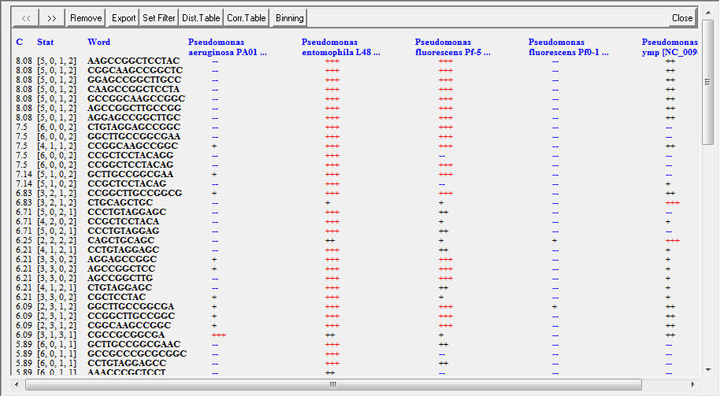
Description of the command buttons on the report panel is given below.
2. Selection of common abundant words.
In this mode the program selects the words which are abundant in all
selected taxa. Repeat the same steps as in the previous task but click the
button Select common abundant words or
choose Command->Select
abundant words.
3. Selection of common rare words.
In this mode the program selects the words which are rare in all selected
taxa. Repeat the same steps as in the first task but click the button
Select common rare words or choose
Command->Select rare
words. The resulted list of words will be shown on the right
panel.
4. Comparison of different taxonomic units.
The program selects words with the minimal divergence within the taxonomic units but
the maximal intrataxonimic divergence. Choose
Command->Compare taxa. A dialog will
pop up shown below:

Select a radio-button to display the list of classes, genera
or species. Then in the scrolled listbox chouse the taxonomic units to
compare. User may click Show/Hide Filter
button to set the word filter (will be discussed below), but at this stage
it is not recommended to use the filter as it will significantly slow down
the program.
Click OK. The resulted list of top
scored words
will be shown on the right panel.
5. Confronted comparison of taxonomic units.
In this mode the program selects the words, which will be the most useful to
distinguish a selected taxon from a set of counterpart taxonomic units; or
the words which are abundant in the selected taxon but rare in the counterpart taxa; or contrary, the words which are rare in the selected taxon but
abundant in the counterpart taxa.
Choose Command->Confronted
comparison. A dialog will pop up shown below:
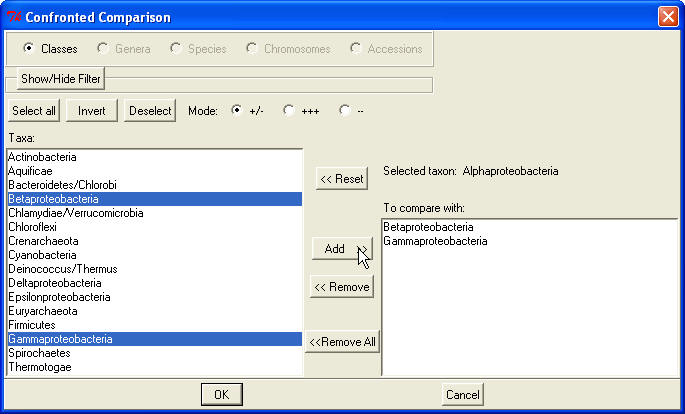
Select the radio-button to display the list of classes,
genera, species or chromosomes. Choose the radio-button
+/-, +++
or -- to select the divergent, abundant
or rare word selection, correspondingly. Then in the scrolled listbox select the
sample taxonomic unit and click the button Select.
The name of the taxon will appear in the area
Selected taxon, the button Select
turns to Reset and the listbox will
allow multiple selection of taxonomic units. Now select the taxonomic units
against which you want to compare the sample taxon and click the button
Add to
display these items in the list To compare with.
User may click Show/Hide Filter button
to set the word filter (will be discussed below), but at this stage it is
not recommended to use the filter as it will significantly slow down the
program.
Click OK. The resulted list of words
will be shown on the right panel.
▪ << – shift the result table leftward for one screen;
▪ >> – shift the result table rightward for one screen;
These two commands are used if the result table contains too many columns. Five columns are displayed at a time. If more genomes are selected, used the buttons << and >> to navigate around the wider table.
▪ Remove – open Remove genomes or words. Remember, that when genomes are removing using this dialog, the scores of the selected words will not be updated! To select words for a smaller set of genomes, select the required genomes and recalculate the table.
▪ Export – export the result table or the list of top scored signature words to a text editor that allows saving the table as a text file. The saved report file may be imported back to the Report panel using the menu command File->Import report.
▪ Set Filter – open Word filter dialog. This option will be explain in the next section.
▪ Dist.Table – calculate distance table for selected genomes. This option is available only for Diverse words report.
▪ Corr.Table – calculate correlation table for selected genomes. This option is available only for Diverse words report.
▪ Binning – calculate distances between an unknown sequence or a group of sequences stored in a FASTA file and the selected genomes or taxonomic units based on counting the signature words listed on the Report panel. Click the button Binning and select the input FASTA file in the Open file dialog. Distance values in the range from 0 (identity) to 10 (maximal distance) will be assigned to each selected genome or taxonomic unit.

Filter settings may be done prior to running the word selection
algorithms described above, but we recommend not to set filtering of word
permutations, wordshifts and constituent words at this stage, as it may make
the program too slow.
More practical will be filtering of the words in the resulted list. Not only
it is faster, but word filtering in this case is reversible.
To open Word filter dialog, click on the
button Set Filter. Following options are
available in the dialog:
▪
Filter permutations – click on the check
button to activate the filter and select the value in the combo box. The
program will remove from the list all words with lower scores, which may be
obtained from the outscored word by one or several nucleotide replacements;
▪ Filter wordshifts
– click on the check button to activate the filter and select the value in
the combo box. The program will remove from the list all words with lower
scores, which may be obtained by shifting the outscored word to one or two
nucleotides leftward or rightward;
▪ Filter
constituents – click on the check button to activate the filter
and select the value in the combo box. The program will remove from the list
all words that are constituent elements of longer words already present in
the list;
▪ Word length
thresholds – set the minimal and the maximal length of the words
in the range from 8 to 14 nucleotides;
▪ Score –
in all algorithms the words are scored in the range from 0 to 10. Set the
minimal value of the score for the words to be selected;
▪ Top –
maximal length of the list of selected words.
▪
Redun –
maximal number of redundant words.
Look at the table above. Words 1 and 2 are highly discriminative
but the word W2 doesn't add any new information to differentiate 6
selected genomes. Option Redun by
default is set to 10, that means that only 10 words with identical (or
similar, see below) distribution of percentiles are allowed in the list. It
gives a way for other discriminative words, such as W4, even if they have
smaller scores.
G1
G2 G3 G4
G5
G6 W1 +++ +++
+++ -- --
-- W2 +++ +++
+++ -- --
-- W3 +++ +++
++ -- --
-- W4 ++ --
+++ --
++ --
▪ Sim –
minimal dissimilarity in percentile distribution to consider a word not to
be redundant to another word. Look at the table above. The word W3 is
distributed very much similar to the words W1 and W2 except for the genome
G3. The difference between neighbour percentiles is counted as 1/3. If word1
is abundant (+++) in a genome and word2 (--) is rare in the same genome, it
will add 1 to the difference between words. Correspondingly, the maximal
difference between two words may be equal to the number of selected genomes
- 6 in our example. The difference between W2 and W3 is 1/3 that is
approximately 5.5% of the maximal difference. By default the option
Sim is set to 10%, thus under this
setting the words W1, W2 and W3 will be considered by the program as
redundant.
To reverse word filtering, click again on the button
Set Filter and click
OK without setting any values.
To check which words are in the database or to display frequencies of the specific words in different genomes, use the command File->Search words. A dialog will pop up that is shown below:

Enter an oligonucleotide of 8 to 14 bp long into the field
Search for and click on the button
>> to add this oligonucleotide to the
list Selected words. Alternatively, you
may enter only a few starting letters (nucleotides) of the word and click the button
Search. A list of all words in the
database starting with the entered letters will appear in the listbox
Available words. Click on the word of
interest and then click on the button >>
to add this oligonucleotide to the list Selected
words.
Words may be uploaded to the list Selected words
from a text file using the button Upload.
The list may be created manually or exported from the
Report panel using the button Export.
Next select taxonomic units in the listbox Selected
genomes and click on the button OK.
The program will calculate either the discrimination scores for selected
words and taxonomic units, if the radio button Taxa
is set; or displays frequencies of the selected words as shown below, if the
radio button Words is set:


To open the current help file in the browser on your computer, choose the command Help->Help.
python oligodb_cmd.py -i
input.txt -d bacteria -o output.out
-w words.txt -p 0 -f
0,0,0,8,14,0,10000,10,10
▪
-i –
the name of the input file with the list of NC accessions for which the
discriminative signature words are to be found (input
by default). The input file has to be
specifically formatted. The file may be obtained by selecting the genomes
or taxonomic units of interest in GUI main window and exporting their
accession numbers to a text file using the command
File->Export
accessions. Alternatively, use the
command line program infile_cmd.py
that is described below.
▪ -d –
the name of the database (bacteria by
default). It is supposed that the database file with the
same name and the extension .wdb
is in the subfolder db of the current
folder.
▪ -o –
the name of the output report file
(output by default). The file format will be exactly the
same as when the button Export on the
Report panel is used (see above).
Later this file may be displayed on the
Report panel using the command File->Import
report.
▪ -w –
optional name of a text file with the list of pre-selected words. By
default this option is not set. It is
similar to uploading the word list file in the dialog
Search words and running the program
for the taxa selected in the same dialog with the radio button
Taxa set on (see above).
▪ -p –
program number (by default 0):
0 – select diverse words or compare
taxa if accessions are grouped by taxa in the input file;
1 – select abundant words for genomes
or taxa;
2 – select rare words for genomes or
taxa;
3 – do confronted comparison. First
genome or first taxa in the list are selected as an outgroup.
▪ -f –
the string of filter settings in the order "m,sh,c,wl1,wl2,sc,top,red,sim"
where:
m – number of permutations to filter
(may be set as an absolute number or as a percentage);
sh – left/right shift length to filter
(may be set as an absolute number or as a percentage);
c – length of constituent words to
filter;
wl1 – minimal word length (in a range
from 8 to 14 but smaller than wl2 );
wl2 – maximal word length (in a range
from 8 to 14 but bigger than wl1);
sc – score threshold;
top – number of top scored words to
select (by default 10000);
red – maximal number of redundant
words in the list (by default 10);
sim – similarity threshold to consider
words as redundant (by default 10%, see above);
This program is used to prepare an input file
for the program oligpdb_cmd.py that
was described above.
python infile_cmd.py -t
2 -d
bacteria -o output.out -n
NC_009725,NC_003997,...
▪
-n –
comma separated list of accessions or taxonomic units;
▪
-t –
taxonomic level (2 by default):
0 – bacterial classes:
-n
Firmicutes,Fusobacteria,... ;
1 – genera:
-n
Firmicutes|Bacillus,Firmicutes|Clostridium,...;
2 – accessions:
-n
NC_009725,NC_003997,...;
3 – species:
-n
Firmicutes|Bacillus|subtilis,Firmicutes|Bacillus|cereus,....
▪ -o –
output file that may be used as an input file for the program
oligpdb_cmd.py.
Selection of high scored words and filtering
of the word list may be separated to achieve better performance. Use the
program filter_cmd.py to filter the
output file of the program oligpdb_cmd.py
with the word list report.
python filter_cmd.py -i
report.txt -o
filtered_report.txt -f
30%,1,1,8,14,0,100,10,10%
▪
-i –
name of the input report file that is to be filtered;
▪
-o –
name of the output file;
▪ -f –
the string of filter settings in the order "m,sh,c,wl1,wl2,sc,top,red,sim"
where:
m – number of permutations to filter
(may be set as an absolute number or as a percentage, by default
30%);
sh – left/right shift length to filter
(may be set as an absolute number or as a percentage, by default
1);
c – length of constituent words to
filter (by default 1);
wl1 – minimal word length (in a range
from 8 to 14 but smaller than wl2 );
wl2 – maximal word length (in a range
from 8 to 14 but bigger than wl1);
sc – score threshold;
top – number of top scored words to
select (by default 100);
red – maximal number of redundant
words in the list (by default 10);
sim – similarity threshold to consider
words as redundant (by default 10%, see above);
The database may be updated with new genomes
represented by FASTA or GenBank files.
python
dbupdate_cmd.py -i
input -d
bacteria
▪
-i –
name of the input folder - input by
default;
▪
-d –
the name of the database (bacteria by
default). It is supposed that the database file with the
same name and the extension .wdb
is in the subfolder db of the current
folder.
Before running this program, copy source genbank
files to the input directory. In contrast to the function Edit->Add
that may parse both FASTA and GenBank file formats,
this command line program can read only GenBank files.