SeqWord Genomic Island Sniffer |
Download
|
NEW: GI Viewer |
SeqWord Sniffer GI BrowserLingvoCom |
| This program was developed to allow an automatic search in genomic DNA sequences for loci enriched with putative horizontally transferred elements, fitness genes, giant genes, or genes for ribosomal RNA and proteins. Predictions are made using oligonucleotide signatures of the genomic fragments. | |
| Installation Options for the run and preset scenario Input and output files Options to improve prediction Editing the task list Setting task conditions Addition of a new task Save new scenario Setting the size of the sliding window Input and output folders Publications |
|
| Installation | The program needs no installation. Download
SeqWordSniffer.zip.
The file contains a Python version of the program compatible with all OS
with Python 2.5 installed. Unzip file to a selected
directory. A folder SeqWordSniffer will appear with several files inside
and two subordinate folders input and output. To process genomic DNA
sequences in FASTA or GenBank formats copy them to the folder input and
run the file SeqWordSniffer.exe or python SeqWordSniffer.py depending on
the version of the program you have got. A console window will appear
(examples of the Command Prompt window in MS Windows are shown below): |
| Options for the run and the preset scenario | The window shows the run options set
by default. Several sets of options were created and stored as scenarios
to identify the loci of interest in a genomic DNA sequence prior to
annotation. The identification is based on the analysis of oligonucleotide
usage (OU) statistical parameters as described in our previous
publications [1, 2]. By default the options are set to identify
horizontally transferred genomic elements. To change the scenario , press
<C> + <Enter> and
select a new scenario by its number: |
| Input and output files | To run the program press <Y> + <Enter>. The
program sequentially processes all files of genomic DNA sequences in FASTA
('FNA','FAS','FST','FASTA') or GenBank ("GBK","GB") formats from the folder input and
saves the results as in the folder output. Several types of output
files may be saved:
Text output file The output files contain information about all genomic fragments enriched with the genes of interest as in the following example: <GI> NC_013209:1 <COORDINATES>
187675-206574 <STAT> n1_4mer:GRV/n1_4mer:RV = 2.318036; n0_4mer:D =
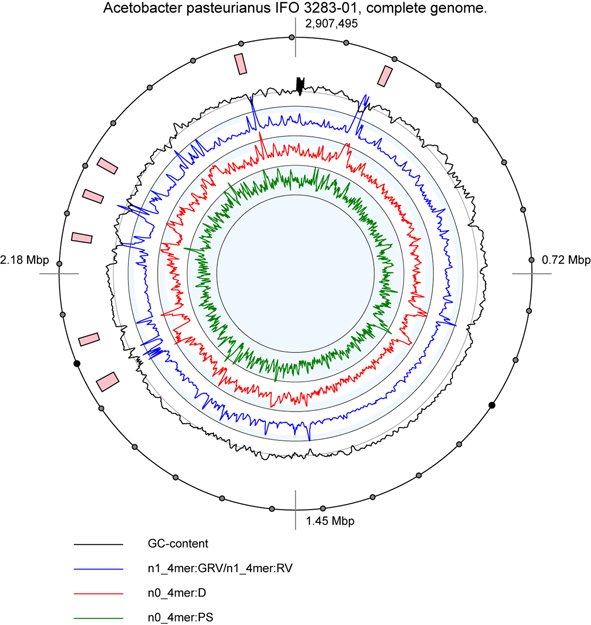
39.633998; n0_4mer:PS = 32.759776 Graphical output file
An example of the graphical output file. Pink blocks show positions of predicted genomic loci of interest.
|
| Options to improve prediction | There several options are available which may improve the
prediction of the loci of interest but at the expense of the program run
time.
|
| Edition of the task list | The user may change the default
options. To change the set of the OU statistical parameters the program
calculates to identify the genomic fragments press <T> + <Enter>: Each task is presented by a line defining the task category and the condition used to select the genomic fragments. Remember that the fragment will be selected only if it meets all set conditions. To remove a condition press <R> + <Enter>, then select the number of the task to remove it from the list. To return to the main menu press <Q> + <Enter>. |
| Setting task conditions | To edit the condition of one of the
tasks press <E> + <Enter>. Now type the number of the task to edit and press <Enter>. A submenu of edit options will appear as shown below: Use the option <M> to choose the type of the threshold values:
To choose the type of comparison,- bigger than,
smaller then or between, - press the key <G>,
<S> or
<B>
respectively and press <Enter>. The program will prompt to enter the
values of one or two (if the option Between is used) thresholds. To choose
values of thresholds consult the SeqWord Browser program
(http://seqword.bi.up.ac.za//mhhapplet.php) as in the examples below: |
| Addition of a new task | To add a new task press <A>+ <Enter>. The
program will show a new menu:
Press <A>+<Enter> to add a subtrahend
or a divisor, or to add the new task to the list. In the letter case the
program will show the condition setting menu that was described above.
Press <Q>+<Enter> to return to the task edit menu and again
<Q>+<Enter> to return to the main menu. |
| Save a new scenario | If the list of tasks is changed, the program changes the
name of the current scenario to "User defined". To save the new
list of tasks in the main menu press <A>+<Enter> and name your
scenario. |
| Setting the size of the sliding window | The program identifies gene islands by using a sliding
window approach. To achieve optimal speed and accuracy of identification
of gene islands the program flexibly changes the step of the sliding
window choosing between big, medium and small steps (see below): To change the values of the sliding window length (8 000 bp), big step (2 000 bp), medium step (500 bp) and small step (100 bp) set by default, press the keys <L>, <B>, <M> and <S> correspondingly and press <Enter>. The program will prompt you to enter new values. (Remember that for statistical reliability the sliding window size should not be shorter than 4600 bp for tetranucleotide usage analysis, 1200 bp for trinucleotides and 600 bp for dinucleotides. |
| Input and output folders | By default the program reads sequence files from the folder
input and saves the result files (see an example above) to the folder
output. A user may change names of the input and output folders from the
main menu by selecting the options <I> and <O>. In addition to
the text files with coordinates of identified gene islands it is possible
to instruct the program to save the sequences of the gene islands to FASTA
files. To do this press <F>+<Enter>. |
| Publications |
|