Identification
of horizontally transferred genomic islands
Genomic islands can be located due to their divergent tetranucleotide usage compare to the core genome. A key feature of the SeqWord Genome Browser is the use of three distinct parameters to identify genomic islands, instead of simply the traditional GC alteration.
To select all genomic islands of a bacterial genome, go to the tab ‘Diagram’.
Select n1_4mer:RV for the X axis, n1_4mer:GRV for the Y axis and n0_4mer:D for the Z axis.
Click the button ‘Enter’.
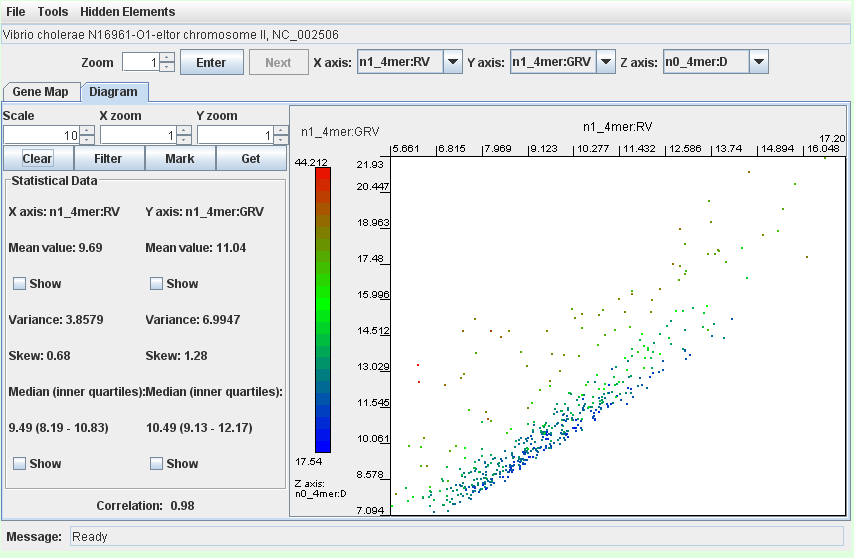
Genomic fragments that correspond to putatively horizontally transferred elements are presented in a cloud of green, brown or red dots above the lower part of the mainstream diagonal distribution of the core genome fragments. Select the dots as shown on the image and click the button ‘Get’.
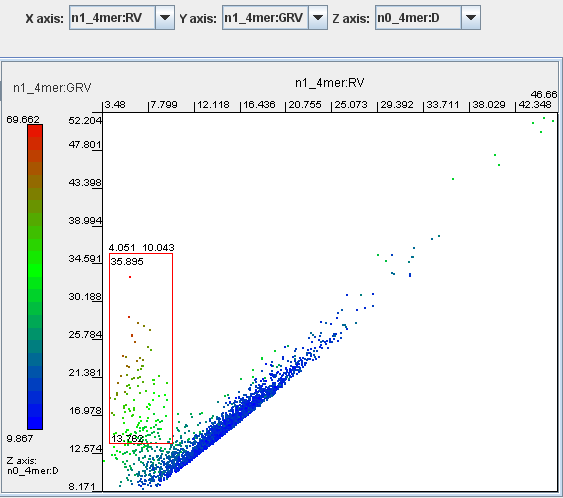
The above image is of Pseudomonas putida KT2440, which as a known mosaic genome contains many genomic fragments dissimilar to the genomic average.
For more precise identification use the filter with the sliders set as shown:
Warning - the vertical layout of the parameters may change according to how many parameters are calculated for the sequence.
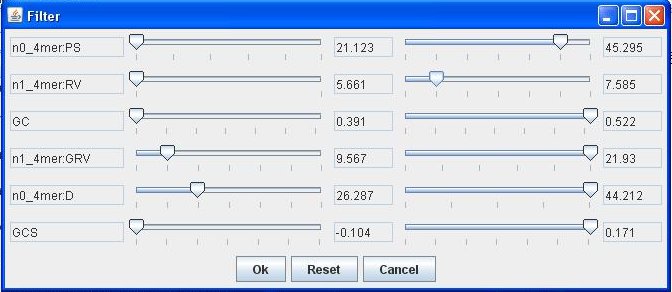
Having set the filter, only fragments that correspond to gene islands remain on the dot-plot:

This method has been successfully demonstrated for several strains with known genomic islands:
- the SKIN element in Bacillus subtilis 168
- phage related genomic islands in P. putida KT2440 and in Salmonella enterica Ty2
- the LEE pathogenicity island in E. coli O157:H7
- IS-elements, pathogenicity islands and prophage regions in Shigella flexneri 2457T
- the ISFtu1 element in Francisella tularensis Schu 4
- the cag pathogenicity island in Helicobacter pylori 26695
- the 67 kbp genomic island in X. fastidiosa 9a5c.
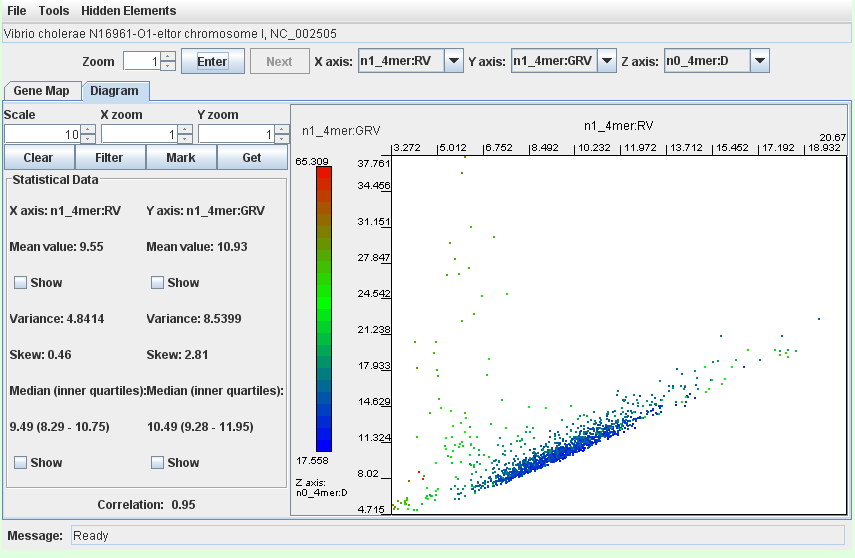
- the integron island in Vibrio cholerae N16961-O1-eltor chromosome II (worked example)
- the large 680kb symbiotic island in Bradyrhizobium japonicum USDA110
All above mentioned genomic islands were successfully localized following comparison of local and global OU patterns.
However, not all islands are quite so divergent. For example, some islands may have been gained from a location compositionally similar to the current genome (for example, from a strain with similar GC content and tetranucleotide usage). These islands are thus more difficult to detect.
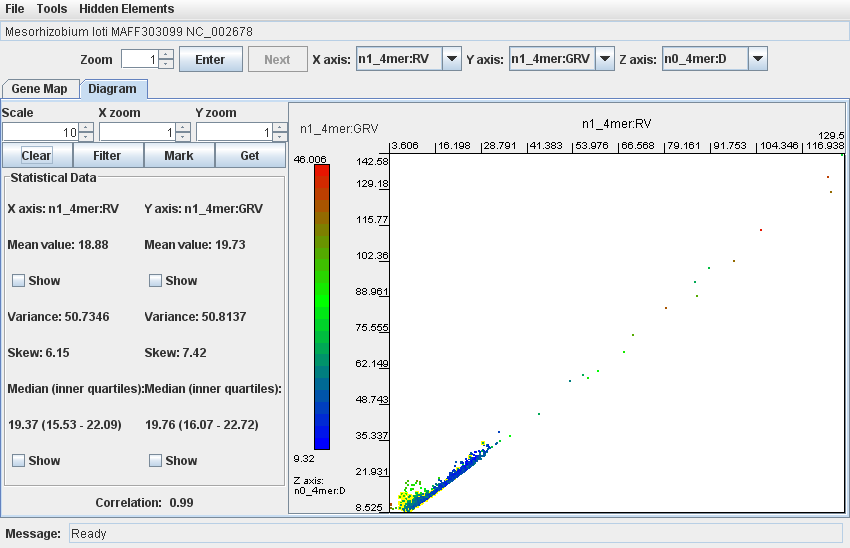
For example, Mesorhizobium loti MAFF303099 contains a large symbiotic island (similarly to B. japonicum USDA110). The island has an average GC content of 59.4 % compared to the genomic average of 64 %, and as such may have been partially ameliorated (where the island GC and OU patterns become more similar to the host OU over time due to accumulations of mutations in especially redundant codon positions. Whether ameriolation has taken place or not, this island is difficult to detect with the current protocol. The below screenshot shows a dotplot of this strain. Fragments within the island coordinates (here 4.70MB to 5.20MB) have been coloured yellow with the "Mark" button, and are not distinct enough from the core genome (coloured blue) to be reliably detected.