LingvoCom 1.0
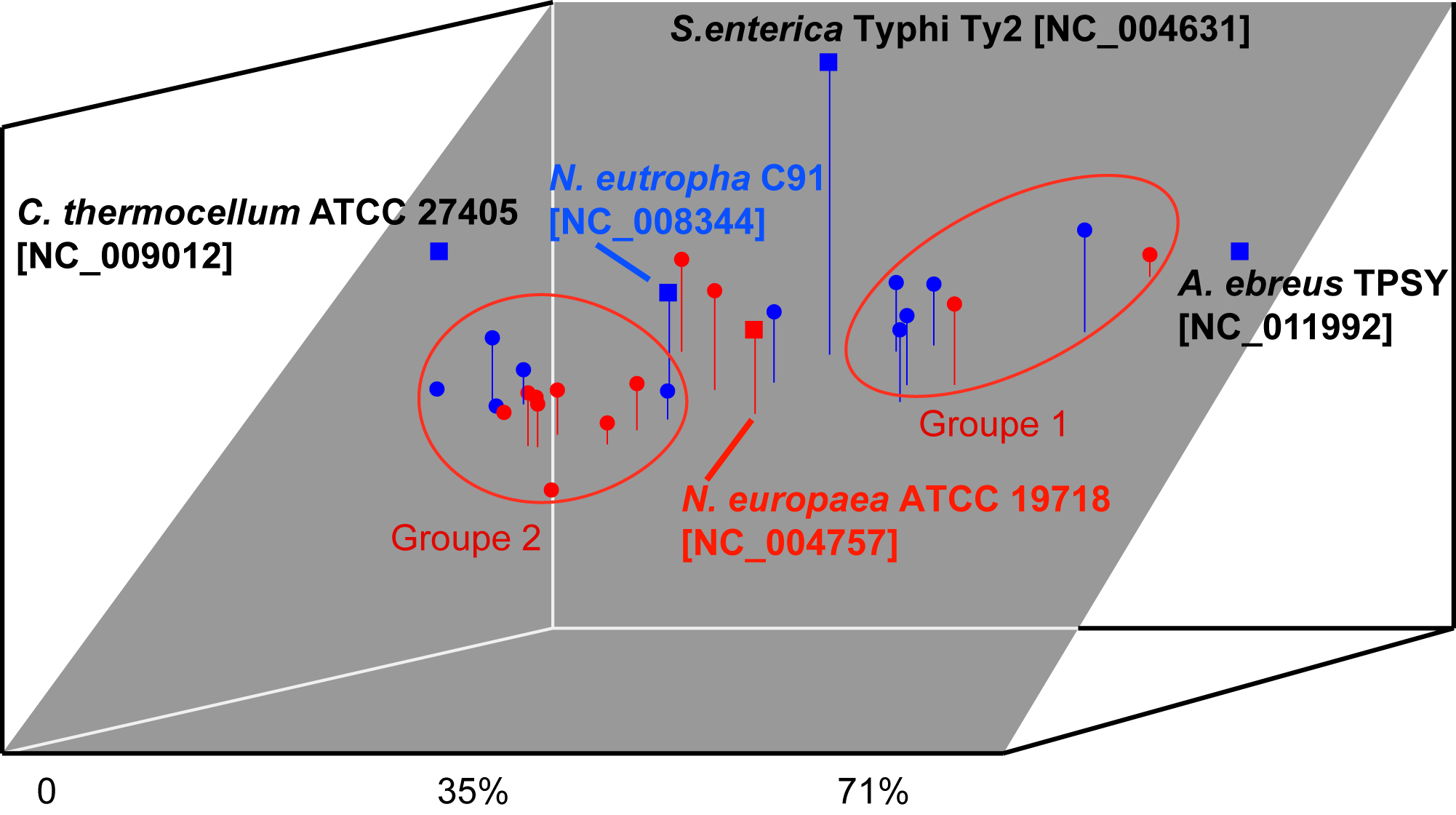
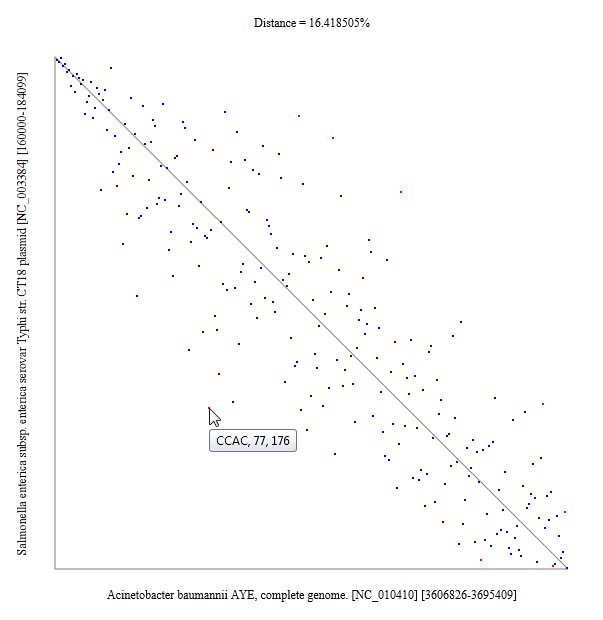
Oligonucleotide usage pattern comparison and visualization
Program download
Last update: April 25, 2012
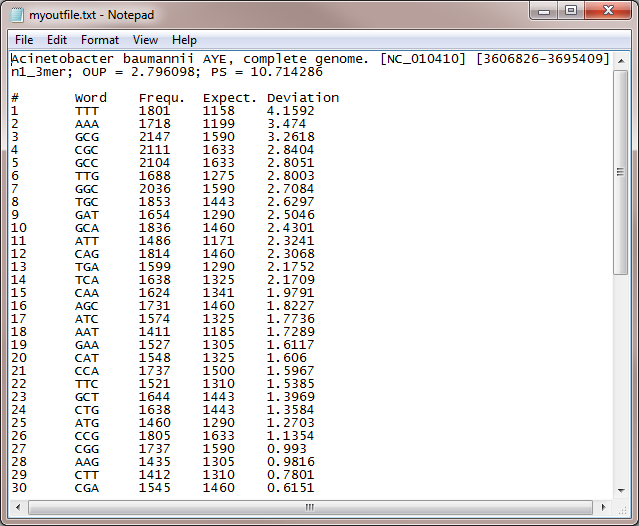
The program LingvoCom is written in Python and requires Python ver. 2.5 to be installed on the machine. The program works with DNA sequences in FASTA or GenBank formats. Visualizations are saved as vector graphic SVG files, which may be viewed by Mozilla Firefox or Chrome browsers, or vector graphic editors such as Adobe Illustrator.
-
To run the program on a Windows machine, double click the file lingvocom.py and a command prompt window will appear:
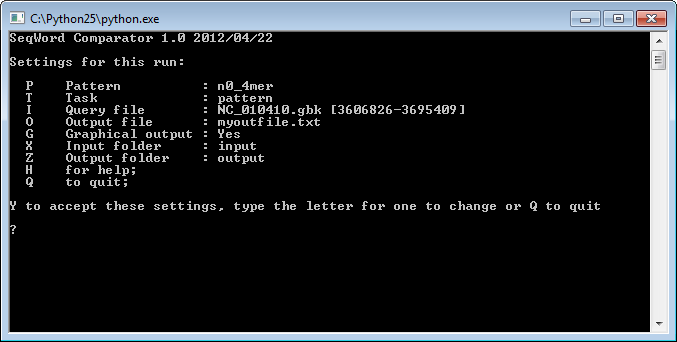
to run the program, type <Y>+<Enter>; -
In Linux type the command python lingvocom.py;
-
Alternatively, the program may be run with a list of arguments:
lyngvocom.py -p n0_4mer -t pattern -i "NC_010410.gbk [3606826-3695409]" -o myoutfile.txt -g Yes -x input -z output
When run like this, all 7 arguments must be set, otherwise a prompt menu will be shown.
In the prompt menu to change the default setting for an argument, call it by typing the corresponding letter + <Enter>, then type or chose an alternative value.