How to run the applet locally
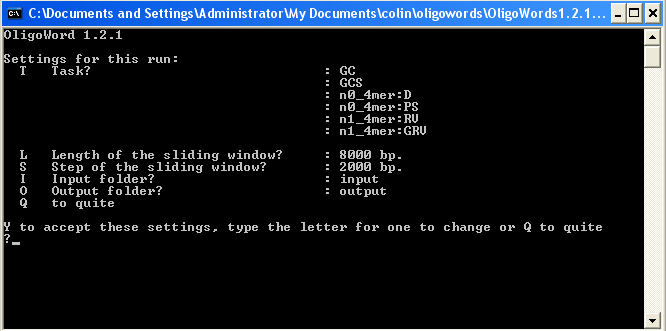
To change the task list, type 't' and press 'Enter'. New menu will appear that allows you to add or remove tasks from the list. Type 'a' to add a new task and then type 'c' to select task category:
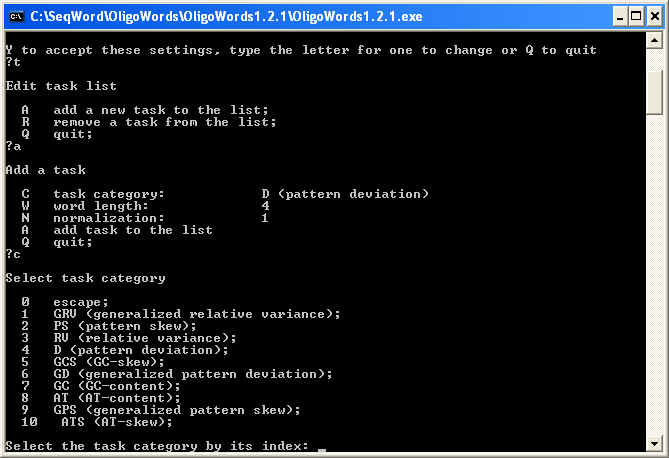
One may select following task categories:
*generalized means that the expected frequency of nucleotides will be normalized by frequencies of constutuent oligonucleotides calculated for the complete genome rather than for a given sliding window (default).
Next select the scheme of normalization ('n') and the word length ('w'),
and type 'a', 'q' and 'q' followed by pressing 'Enter'
in three subsequent menus. You'll end up with the
first menu (see above). Type 'y' then Enter to start OligoWords.
OligoWords will process the fasta/Genbank files in the input directory. The output
is plain text files with the extension '.out' in the
output directory.
The number of output files will be the same as the number of input sequences.
Note:
to ensure reliability of statistical calculations keep in mind that the sliding
window length should not be shorter than the following limits. These are set in relation to the maximum word length value.
| Word length (bp) | Minimum sliding window size (bp) |
| 2 | 300 |
| 3 | 1200 |
| 4 | 5000 |
| 5 | 18500 |
| 6 | 74000 |
| 7 | 295000 |
If the word length set for one task requires a larger sliding window, the
program will perform calculations as it is set without any warnings, but the
resulting values will be statistically unreliable!
2 Viewing output files with the SeqWord Genome Browser locally
To
view an output file use the on-line version of the SeqWord Genome Browser or a
local version of the applet MhhViewer.html (with associated Java jar file) which is freely available for download.
If you use the local applet, open it with your webbrowser and
allow the browser to display the applet by accepting the certificate if prompted.
Select File > Open and navigate to the OligoWords output folder to select the results of interest (image below)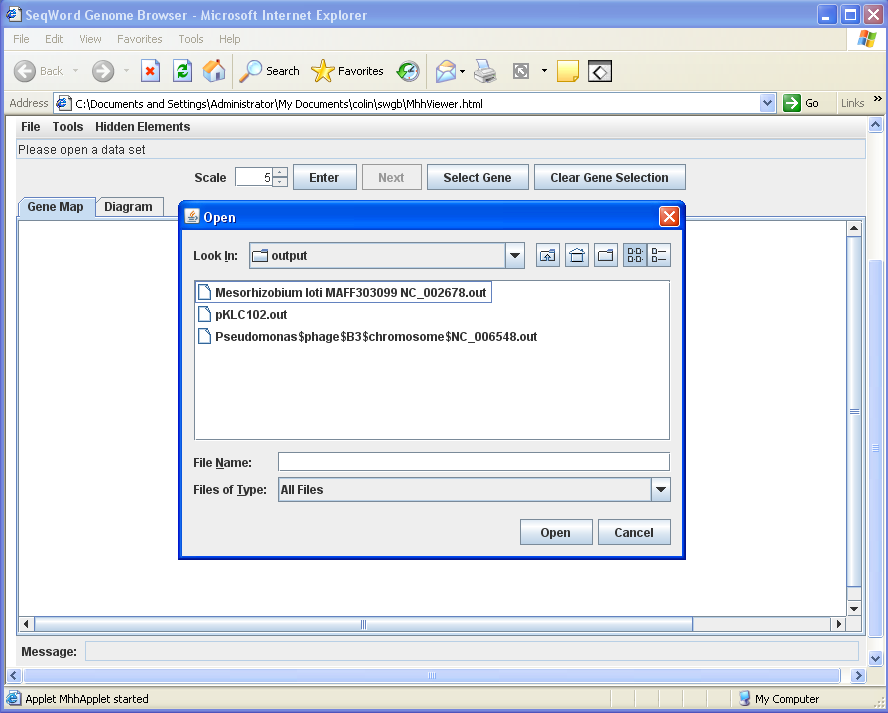
Now you can view and analyse your results just like in the server version of the SeqWord Genome Browser.
Gene
names and positions are of course not provided if you only use a fasta
sequence, since no annotation exists for the genome with this file type.